10月31日,江西华星技术检测有限公司与宜春学院化生学院签订校企合作战略协议,为双方开展实习基地创建、科研项目合作、校企资源共享、学生招聘就业等方面打下了坚实的基础。



 微信服务号
微信服务号点击咨询 全国咨询热线:: 152-7000-1226
index.php
content
10月31日,江西华星技术检测有限公司与宜春学院化生学院签订校企合作战略协议,为双方开展实习基地创建、科研项目合作、校企资源共享、学生招聘就业等方面打下了坚实的基础。
content
为进一步加强食品安全监管,确保人民群众“舌尖上的安全”,10月28日开始,樟树市市场监督管理局启动2019年度食品安全抽检工作,其中样品检测工作由华星检测全面负责,助力樟树市管局打击市场上生产、流通过程中不合格食品,为樟树市人民群众食品安全保驾护航。
此处食品安全抽检工作覆盖了粮食加工品、肉制品、面制品、餐饮食品等20多个品类,近2000个批次。华星检测将秉承“科学、公正、高效、精准”的态度,全程参与食品抽检工作的每个环节,全力将检测服务工作做到极致。


content
经过一个多月紧锣密鼓的筹备,江西华星检测网站终于开通上线了!
content
本汇总由食品伙伴网合规事业部整理,主要是食品行业常用标准,包括:通用基础标准:GB 2761-2017 食品安全国家标准 食品中真菌毒素限量、GB 2762-2017 食品安全国家标准 食品中污染物限量等;GB 4789系列食品卫生微生物学检验标准;GB 5009系列食品卫生检验方法理化标准;GB 31621-2014 食品安全国家标准 食品经营过程卫生规范;GB/T 19001-2008 质量管理体系 要求等一些常用标准。
食品安全国家标准-通用标准12项
GB 7718-2011 食品安全国家标准 预包装食品标签通则
GB 28050-2011 食品安全国家标准 预包装食品营养标签通则
GB 13432-2013 食品安全国家标准 预包装特殊膳食用食品标签
GB 29924-2013 食品安全国家标准 食品添加剂标识通则
GB 2760-2014 食品安全国家标准 食品添加剂使用标准
GB 2761-2017 食品安全国家标准 食品中真菌毒素限量
GB 2762-2017 食品安全国家标准 食品中污染物限量
GB 2763-2019 食品安全国家标准 食品中农药最大残留限量
GB 31650-2019 食品安全国家标准 食品中兽药最大残留限量
GB 29921-2013 食品安全国家标准 食品中致病菌限量
GB 14880-2012 食品安全国家标准 食品营养强化剂使用标准
GB 9685-2016 食品安全国家标准 食品接触材料及制品用添加剂使用标准
行业、专业通用标准
GB 30616-2020 食品安全国家标准 食品用香精
GB 29938-2020 食品安全国家标准 食品用香料通则
GB 26687-2011 食品安全国家标准 复配食品添加剂通则
GB 14881-2013 食品安全国家标准 食品生产通用卫生规范
GB 31621-2014 食品安全国家标准 食品经营过程卫生规范
GB 31654-2021 食品安全国家标准 餐饮服务通用卫生规范
GB 31605-2020 食品安全国家标准 食品冷链物流卫生规范
JJF 1070-2005 定量包装商品净含量计量检验规则
GB/T 1.1-2020 标准化工作导则 第1部分:标准的结构和编写
GB/T 19001-2008 质量管理体系 要求
GB/T 22000-2006 食品安全管理体系 食品链中各类组织的要求
GB/T 22004-2007 食品安全管理体系 食品安全管理体系
GB/T 27341-2009 危害分析与关键控制点(HACCP)体系 食品生产企业通用要求
GB/T 191-2008 包装储运图示标志
GB/T 6543-2008 运输包装用单瓦楞纸箱和双瓦楞纸箱
GB/Z 21922-2008 食品营养成分基本术语
检验检测相关
GB 14882-1994 食品中放射性物质限制浓度标准GB/T 27025-2019 检测和校准实验室能力的通用要求
GB/T 27404-2008 实验室质量控制规范 食品理化检测
GB/T 27405-2008 实验室质量控制规范 食品微生物检测
食品卫生微生物学检验标准-4789【标准专题】
食品卫生检验方法理化标准-5009【标准专题】
GB/T 2828.1-2012 计数抽样检验程序 第1部分:按接收质量限(AQL)检索的逐批检验抽样计划
GB 5749-2006 生活饮用水卫生标准
GB/T 5750 生活饮用水标准检验方法(GB/T 5750.1-2006~GB/T 5750.13-2006)
GB/T 6682-2008 分析实验室用水规格和试验方法
GB/T 601-2016 化学试剂 标准滴定溶液的制备
24种标准滴定溶液:氢氧化钠、盐酸、硫酸、碳酸钠、重铬酸钾、硫代硫酸钠、溴、溴酸钾、碘、碘酸钾、、草酸、高锰酸钾、硫酸亚铁铵、硫酸铈(或硫酸铈铵)、乙二胺四乙酸二钠、氯化锌、氯化镁(或硫酸镁)、硝酸铅、氯化钠、硫氰酸钠(或硫氰酸钾或硫氰酸铵)、硝酸银、亚硝酸钠、高氯酸、氢氧化钾-乙醇
GB/T 602-2002 化学试剂 杂质测定用标准溶液的制备
GB/T 603-2002 化学试剂 试验方法中所用制剂及制品的制备
HG/T 3696.1-2011 无机化工产品 化学分析用标准溶液、制剂及制品的制备 第1部分:标准滴定溶液的制备
26种标准滴定溶液:氢氧化钠、盐酸、硫酸、碳酸钠、重铬酸钾、硫代硫酸钠、溴、溴酸钾、碘、碘酸钾、、草酸、高锰酸钾、硫酸亚铁铵、硫酸铈(或硫酸铈铵)、乙二胺四乙酸
content
色谱分析过程中,缓冲液是不可或缺的,因为要用它来调节流动相的pH值,缓冲盐使用不当,后果可是吃不了兜着走。
比如说,色谱柱压升高、柱效下降、保留时间异常等等不良影响:
1)柱压升高;
原因:缓冲盐使用不当导致缓冲盐析出,堵塞塞板和键合相颗粒之间的孔隙,阻碍流动相传质,引起柱压升高。
2)相同化合物的保留时间发生变化;
原因:如果没有冲洗干净就进行进样,色谱柱内含有的盐会使化合物的保留时间发生变化.
3)柱效下降;
原因:i)有些缓冲盐会渗入到键合相的深处,损害硅胶基体,导致色谱柱键合相流失,柱床变松,柱效下降;ii)凝结在键合相表面,使C18碳链难以舒展,对物质的保留能力下降,导致柱效下降。因此用过缓冲盐后需要对色谱柱进行冲洗,水中缓冲盐浓度较大时应特别引起注意。
使用前和使用后的处理都是有讲究的哦!
1. 使用前的处理: 在使用缓冲盐作流动相之前需要用不含缓冲盐的流动相冲洗色谱柱,直至基线平稳。 原则上,用于冲洗的流动相与分析时所用的流动相含水的比例相同(或含水更多),不同的只是用于冲洗用的流动相中不含缓冲盐。理由:缓冲盐通常易溶于水,难溶于有机溶剂。用含缓冲盐的(特别是做流动相的水为饱和的缓冲盐溶液时)流动相进行分析时,如果分析前色谱柱中用于保存色谱柱的流动相中含水的比例相对较小,不先冲洗掉,接下来做样品的时候所用的流动相中如果有机溶剂含量大,而其比例中所含的水又不足以溶解该缓冲盐时,缓冲盐将会在色谱柱柱体上析出,沉积下来,这将可能导致上述对色谱柱的损害.
2. 使用后的处理:用与分析时含水比例相同的流动相(与分析用流动相唯一的区别是,用于冲洗的流动 相不含缓冲盐)进行冲洗约30min,直至基线平稳。如果该色谱柱在接下来很长的一段时间内不使用,要长期保存,则需再加上一步,即用纯的有机溶剂冲洗一遍,直至基线平稳。
用与分析时含水比例相同的流动相(与分析用流动相唯一的区别是,用于冲洗的流动相不含缓冲盐)进行冲洗约30min,直至基线平稳。如果该色谱柱在接下来很长的一段时间内不使用,要长期保存,则需再加上一步,即用纯的有机溶剂冲洗一遍,直至基线平稳. 缓冲液可以提供离子平衡的反离子,并使流动相保持一定的离子强度和ph值,减少拖尾。
注意事项什么的,是关注的重点!
1:避免使用盐酸盐,盐酸盐对钢质有腐蚀作用。
2:缓冲液最好要现配现用,往往缓冲液是良好的菌类培养液,隔天或放置长时间实验时会有很多怪现象发生。
3:实验后不可用有机溶剂直接过度,有机溶剂会处使盐类析出,造成液路或色谱柱堵塞,可用95:5的水甲醇冲洗。
4:使用缓冲液要及时掌握ph范围,做到胸中有数。
5:清洗液路和柱子时,有温控可加热到30摄氏度易于冲洗。
6:长时间用缓冲溶液要注意观察接头处有无析出,若有白色盐类析出,可考虑一定周期用10%硝酸冲洗一下液路(拆下柱子,走30ml,再用5倍水冲洗)可以避免液路的堵塞。
7:选择缓冲液要用可靠的试剂,避免不纯的盐类造成不必要的麻烦。
如果流动相中有机溶剂的比例很高是不能用来冲洗缓冲盐的,是洗不出来的。通常C18柱先用5%~10%的甲醇冲洗,是可以把缓冲盐冲洗出来的,然后用纯的有机溶剂来保护柱子。最好的方法是使用与流动相相同浓度不含盐的流动相进行清洗。但就是速度慢一些。用水是为了快速替换,一般在15分钟以内最好,且用0.8的流速较好. 如果用纯水冲,容易造成键合的碳链的流失,最好用5%~10%甲醇水溶液冲. 可以用纯水代替流动相中的缓冲液,有机相不变。这样冲洗柱子比较稳妥。
小结
正确使用缓冲盐很有必要,既可以防止缓冲盐析出,也可以达到提高色谱柱使用寿命的目的。我们不妨用一句话来总结它的使用方法:用前要过滤,用后需冲洗。
content
在实验室,经常会接触到甲醇、甲醛、甲酸、过氧化氢、磷酸、硝酸、盐酸、氯仿、氢氧化钾、氢氧化钠等等有毒试剂。这些药品一般具有强刺激性、强氧化性以及强腐蚀性,它们可通过呼吸道、皮肤和消化道进入人体而发生中毒现象。因此,了解化学品中毒后的危害性以及中毒后急救方法至关重要。
呼吸道吸入是化学药品进入体内的最重要的途径,如各种气体、溶剂的蒸气、烟雾和粉尘等经人的呼吸道进入肺部,被肺泡表面所吸收,随血液循环引起中毒。
皮肤、黏膜吸收也可使一些能溶于水或脂肪的化学药品经皮肤吸收,再由血液运输到各器官,引起中毒,这是高沸点化合物入侵的主要途径,所以,皮肤上有伤口时,绝不能操作剧毒药品。
影响化学药品毒害性的因素
化学药品在水中的溶解度越大,其危险性也越大。因为人体内含有大量水分,所以越易溶解于水的化学药品越易被人体吸收;有些化学药品若能溶于脂肪,同样能通过溶解于皮肤表面的脂肪层侵入毛孔或渗入皮肤而引起中毒。化学药品经过皮肤破裂的地方侵入人体,会随血液蔓延全身,加快中毒速度。
固体化学药品的颗粒越小,分散性越好,越易引起中毒,因为颗粒小容易飞扬,容易经呼吸道吸入肺泡、被人体吸收而引起中毒。
液体化学品的挥发性越大,空气中浓度越高,从而越容易从呼吸道侵入人体引起中毒。无色无味者比色浓味烈者难以察觉,隐蔽性更强,更易引起中毒。
化学品中毒的危害有哪些?
化学药品中毒会损伤全身器官,如腐蚀性药物会使皮肤、黏膜、眼睛、气管、肺受严重损伤;许多药物如汞、铅、芳香族有机物会使肝脏受到严重的损害;有些药物会积累在某一器官,造成器官损坏。化学药品对人体的危害主要表现为急性中毒和慢性中毒,小编就为大家介绍一下急性中毒和慢性中毒都有哪些症状。
1、急性中毒
急性中毒是指短时间内受到大剂量的有毒物质的侵蚀而对身体造成损害。
窒息
窒息是指人体的呼吸过程由于某种原因受阻或异常,所产生的全身各器官组织缺氧而引起的组织细胞功能紊乱和形态结构损伤的病理状态。
麻醉作用
由于接触高浓度的某些化学药品导致中枢神经抑制而引起头昏,头晕,头痛或昏迷的症状。
全身中毒
全身中毒是化学药品直接破坏肌体组织而引起。
过敏和刺激作用
重复接触某一化学药品引起过敏,化学药品对皮肤、眼睛及呼吸系统刺激后发生化学反应,引起炎症。
2、慢性中毒
慢性中毒是指毒物在不引起急性中毒的剂量条件下,长期反复进入机体而出现的中毒状态或疾病状态,这种伤害通常是难以治愈的。
致癌作用
致癌性指慢性毒性导致癌症,如砷、石棉、氯甲醚可导致肺癌;苯引起再生障碍性贫血;氯乙烯单体引起肝癌。
诱变和畸形作用
诱变和畸形作用指慢性毒性能够改变细胞基因,威胁正在发育的胎儿,后代的基因会受到损害,使胎儿变成畸形。
具体器官中毒
慢性毒性能够损坏具体器官,如石英晶体、石棉能引起尘肺。
不同途径中毒者的救治
化学药品可以通过呼吸道、皮肤或其他途径对人体造成伤害,引起中毒。那么,不同途径的中毒者要如何进行救治呢?
1、经呼吸道吸入中毒者
首先保持呼吸道畅通,并立即转移至室外,向上风向转移,解开衣领和裤带,呼吸新鲜空气并注意保暖;对休克者应施以人工呼吸,但不要用口对口法,立即送医院急救。
2、经皮肤吸收中毒者
应迅速脱去污染的衣服、鞋袜等,用大量流动清水冲洗15~30min,也可用微温水,禁用热水;头面部受污染时,要注意眼睛的冲洗。
3、误服吞咽中毒者
催吐
适用于神志清醒合作者,禁用于吞强酸、强碱等腐蚀品及汽油、煤油等有机溶剂者。
洗胃
是治疗常规,有催吐禁忌者慎用。
清泻
可通过口服或胃管送入大剂量的泻药,如硫酸镁、硫酸钠等而进行清泻。
content
content
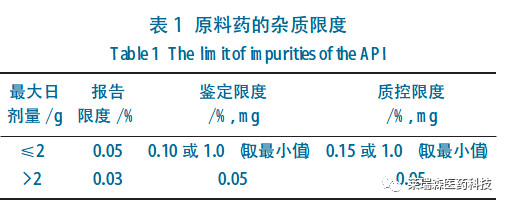
根据《中华人民共和国食品安全法》规定,经食品安全国家标准审评委员会审查通过,现发布《食品安全国家标准 食品中农药最大残留限量》(GB 2763-2021,代替GB 2763-2019)等5项食品安全国家标准。其编号和名称如下:
GB 2763-2021食品安全国家标准 食品中农药最大残留限量
GB 23200.118-2021食品安全国家标准 植物源性食品中单氰胺残留量的测定 液相色谱—质谱联用法
GB 23200.119-2021食品安全国家标准 植物源性食品中沙蚕毒素类农药残留量的测定 气相色谱法
GB 23200.120-2021食品安全国家标准 植物源性食品中甜菜安残留量的测定 液相色谱—质谱联用法
GB 23200.121-2021食品安全国家标准 植物源性食品中331种农药及其代谢物残留量的测定 液相色谱—质谱联用法
以上标准自发布之日起6个月正式实施。标准文本可在中国农产品质量安全网(http://www.aqsc.org)查阅下载,文本内容由农业农村部负责解释。
特此公告。
国家卫生健康委
农业农村部
市场监管总局
2021年3月3日
content
色谱柱的柱效能是评价色谱性能的一项重要指标,混合物能否在色谱柱中得到分离,除取决于选择合适的固定相外,还与色谱操作条件及色谱柱的装填状况等因素有关。在一定的色谱操作条件下,色谱柱的柱效可用理论塔板数或理论塔板高度来衡量。一般说来塔板数愈多,或塔板高度愈小,色谱柱的分离效能愈好。
仪器:高效液相色谱仪(带自动进样器,或配置微量进样器)、分析天平。
试剂:苯、萘、联苯(均为分析纯)、甲醇(色谱纯)、纯净水。
色谱条件:色谱柱:C18,4.6×150mm,5μm;流动相:甲醇-水(80:20,v/v);
检测波长:254nm;流速:1mL·min-1;柱温:30℃;进样量:10μL。
(1)分别精密配制含苯、萘、联苯浓度均为约1mg·mL-1的3份对照品溶液各10mL。
(2)分别精密吸取上述对照品溶液各2mL置于10mL容量瓶中,加流动相稀释,并定容至刻度,摇匀,得到含苯、萘、联苯的混合对照品溶液。
(3)按照上述色谱条件操作,进样,记录色谱图。
(4)计算各色谱峰的理论塔板数及各峰间分离度。
要提高液相色谱的效率可从以下几方面入手。以下介绍了几种国际上流行的测量和计算柱效值的方法。
要提高液相色谱的效率可从以下几方面入手。
(1)降低移动相的流速,但会使分析时间延长。
(2)减少固定相的量,但色谱柱中样品的负载量也随之减小。
(3)减小固定相的颗粒度,但不能过分,过分后色谱柱的渗透率也会减小。
(4)选用低粘度的移动相,以利于快速传质,但却不利于多组份分析。
(5)适当提高柱温,可降低移动相的粘度,但柱效和分离度也随之降低。
(6)尽量减小停滞移动相的体积,但却加快了移动相的流速。
从以上介绍可看出,在色谱分析过程中,各种因素是互相联系和制约的。只有通过对柱效值的跟踪测算,对自己分析方法不断的研究和实践,才能找到最佳的工作条件。
我们也应记住柱效值并不足以预测在所有条件下的柱性能,对大多数色谱工作者来说,柱性能指的是色谱柱用于特定分离的能力,而仅仅有高柱效并不能保证这种分离能力。
不管用什么特定的测试方法,都会有几个参数影响柱效的测定。这些参数包括:洗脱液的成分和粘度及其线流速,测定塔板数所用的溶质,温度,柱长,填料装填方式,颗粒度,还有所选用的测量和计算方法。而测量和计算方法对柱效值的确定起着极大的作用。
因为色谱峰是假定样品浓度在移动相和固定相中呈正态分布而得到的样品谱带分布,故常常把色谱峰型看作正态曲线来计算理论塔板数。因此计算柱效(以理论塔板数n为单位)的公式习惯上定义为:
式中tR为色谱峰的保留时间;σ2是以时间为单位测量色谱峰的偏差;a是和峰高(从测峰宽的基线量起)有关的常数,ωb是峰宽,表示由色谱峰顶点与色谱峰两侧拐点处做切线与峰底基线相交两点间的距离。
假如一个色谱峰真是正态峰型,那么每种计算方法都会得到同样的结果。然而即使一些比较理想的仪器和倾向于得到对称峰型的溶质,由于柱内的槽或空隙,也会出现非正态峰型。所以不同的计算方法将会得到相差较大的n值。通常偏离正态模型的峰型表示为“前延”或“拖尾”。对于这些峰型,越在峰的高处测量,计算的理论塔板数值就越大(准确性越低)。在许多情况下,色谱工作者需要能反映整个峰型(包括拖尾)的柱效值,同时为了保证定量的重复性,也需要色谱峰很好的对称性。这时对色谱峰非对称性最敏感的计算方法最适合。如果目的仅仅是要监测色谱柱从第一次使用到使用寿命结束这一过程中的柱效,那么以上任何一种方法都可以,应选择最简便的方法。
在实际工作中,我们通过对载气流速、进样技术、气化室温度、色谱柱、柱温、检测器温度这六个方面的选择,有效地提高了柱效率,使分析出的色谱峰峰形正常,无峰形扩张、拖尾、峰漏检等不良现象出现,分离度高,从而提高了分析结果的准确性。
所为柱效就是在较短的时间内,用较短的柱子达到满意的分析结果。为了提高色谱柱的柱效率,减少色谱峰扩张、拖尾及峰漏检等现象,在实际工作中,我们从以下六个方面入手,对柱操作条件的选择进行了探讨。
气相色谱最常用的载气是:氢气、氮气、氩气、氦气。
由速率理论可知,载气流速慢有利于传质,有利于组分的分离,但分析时间会加长;如果载气流速快有利于加快分析速度,减少分了扩散,但分离度降低。有时为了缩短分析时间,加大流量,但此时分离效果并不好。可见载气流速的快慢都会降低柱效。经过长时间的实验,发现对于一般色谱仪而言,载气流量为20-100ml/min。目前我们分析液化气用的是热导检测器,载气用的是氢气,其流量控制是30 ml/min。分析戊烷发泡剂用的是氢火焰离子化检测器,载气用的是氮气、燃烧气氢气和氧气,这三种气体的体积比是氮气:氢气:氧气为1:1:10,分析效果都是较好的。
在气相色谱分析中,一般采用注射器或六通阀门进样。在考虑进样技术的时候,以注射器进样为主来研究。
进样量:如果在进样过程中进样量大会导致:分离度小;保留值变化难于定性;峰高和峰面积与进样量不成线性关系,不能定量。进样量与气化温度、柱容量和仪器的线性响应范围等因素有关。进样量应控制在瞬间气化,达到规定分离要求和线性响应的允许范围内。填充柱冲洗法的瞬间进样量:液体样品或固体样品溶液一般为0.01~10μl,气体样品一般为0.11~10ml,在定量分析中,应注意进样量读数准确。
注射器里空气的排除:用微量注射器抽取液体样品,只要重复地把液体抽入注射器又迅速把其排回样品瓶,就可以将空气排除。还有一种更好的方法,那就是用计划注射量的约2倍的样品置换注射器3~5次,每次取到样品后,垂直拿起注射器,针尖朝上,留在注射器里的空气都应当跑到针管顶部,推进注射器塞子,空气就会全部被排掉。
保证进样量的准确:用经置换过的注射器取约计划进样量2倍左右的样品,垂直拿起注射器,针尖朝上,让针穿过一层纱布,这样可用纱布吸收从针尖排出的液体。推进注射器塞子,直到读出所需要的数值。用纱布擦干针尖。至此准确的液体体积已经测得,需要再抽若于空气到注射器里。如果不慎推动柱塞,空气可以保护液体使之不被排走。
进样手法:双手拿注射器。用一只手(通常是左手)扶针插入垫片,注射大体积样品(即气体样品)或柱前压力极高时,要防止从气相色谱仪注样器来的压力把注射器活塞弹出(即用右手的大拇指按压住活塞顶部)。让针尖穿过垫片尽可能深的进入进样口,压下注射器活塞停留1秒钟,然后尽可能快而稳地抽出针尖(抽出的同时继续压住注射器活塞)。
进样时间:进样时间长短对柱效率影响很大。若进样时间过长,遇使色谱区域加宽而降低柱效率。因此,对于冲洗法色谱而言,进样时间越短越好,一般必须小于1秒钟。
气化室温度取决于样品的化学和热稳定性、沸程范围、进样口类型等。合适的气化室温度即能保持样品瞬间完全气化,又不引起样品分解。温度过低,气化速度比较慢,使峰形不规则,出现平头峰或伸舌峰;温度过高使出峰数目变化,产生前延峰,甚至样品分解。为选择合适的气化室温度,在多次的进样中我们发现,气化室温度比柱温高50-100℃或比样品组分中最高沸点高50-70℃较为合适。温度过高过低都会影响柱效。
把柱头变色的填料挖掉,用同类新的填料填充好,效果很好。
正向冲洗色谱柱、反向冲洗色谱柱、超声清洗色谱柱筛板、如果是柱内死体积增加则需更换新柱。
色谱柱是消耗品,理论来说,柱效很低的色谱柱是没得救的,但是有一招,也就是当你发现色谱柱没得救的时候可以用一下,就是反冲色谱柱,但是你要注意的是,流速不要设置太大,这样的话还能再坚持一会。如果这样也没用的话,你就重新买吧。
如果柱子真的很老旧了,就反相冲洗试试,可能还能用一段时间,但不会太久。在然后就只能换填料了,或换新的柱子。
我们前段时间也是柱效不好,其他都比较正常。后来把柱子重新活化了一下,柱效由原来的两千升到四千多了。不过维持了一个月又不行了。